Chapter 1 What is Computational Chemistry?
Computational chemistry is a field of study that takes advantage of the power of computers to generate chemical data. A computational chemist has a very similar, workflow to that of an experimentalist.
While obviously simplified, the main feature in the workflow of a computational chemist is having a computer carry out much of the work to provide chemical information (which I personally consider a major benefit).
1.1 Computable properties
Chemistry can be (somewhat arbitrarly) divided into three categories: structure, equilibrium, and kinetics.(Connors 1990) Computational chemistry has tools to characterize these.
1.1.1 Geometries
What is the lowest energy, ground state geometry of water? General chemistry tells us that water is bent, not linear.

Figure 1.1: Water in a bent and linear geometry.
Below are the relative energies of both these structures at the MP2/aug-cc-pVDZ level of theory.
| Bent | Linear | |
|---|---|---|
| \(\Delta E\) (kcal mol\(^{-1}\)) | -32.8 | 0.0 |
The bent geometry for water is much lower in energy and, therefore, a better model for a ground-state geometry for water.
1.1.2 Orbitals
Molecular orbitals can also be computed and visualized. Below are the HOMO and LUMO for water at the HF/STO-3G level of theory.
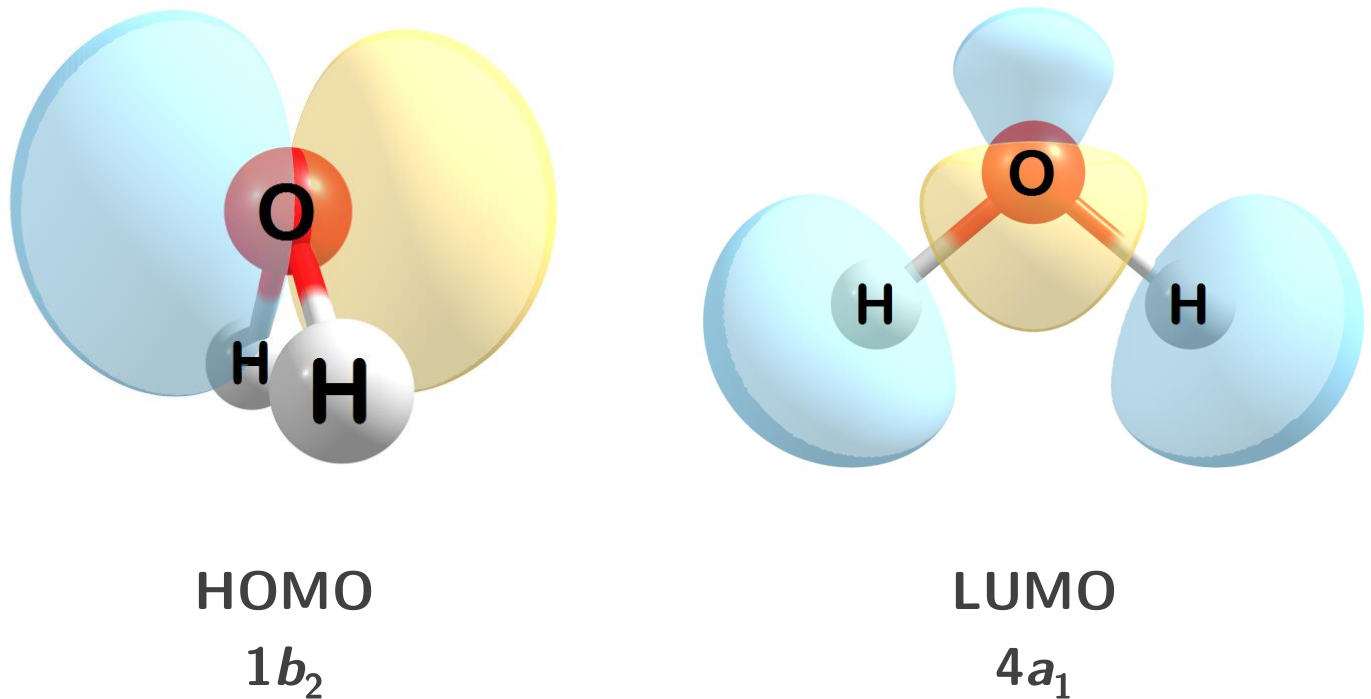
Figure 1.2: The HOMO and LUMO for water.
1.1.3 Charges
Charge distributions, atomic charges, and dipoles can also be computed.
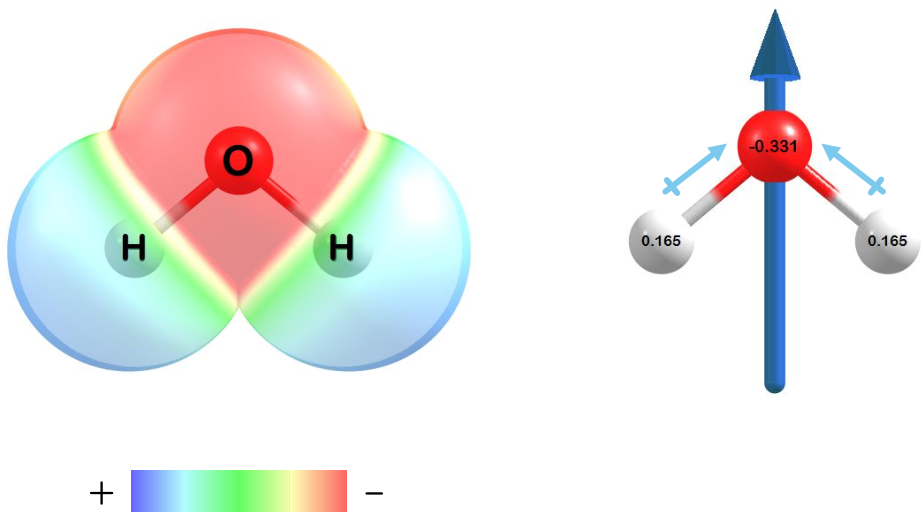
Figure 1.3: The charge distribution (left), atomic charges (right), and dipole moment (right) for water at the HF/STO-3G level of theory.
Electron density in water is concentrated around oxygen (red shaded region) while the hydrogen atoms have a partial positive charge (blue shaded regions). The atomic charges for each atom on water (right) clearly show a partially negative oxygen and partially positive hydrogens. A charge of zero is seen for the molecule when all the partial charges are summed. Finally, the dipole moment (right) points in the direction toward oxygen and opposite that of the hydrogen atoms.
1.1.4 Vibrational modes
Vibrational modes for molecules can also be computed and visualized. Below are the three fundamental vibrational modes (a1, a2, and b1, respectively) for water at the HF/STO-3G level of theory.
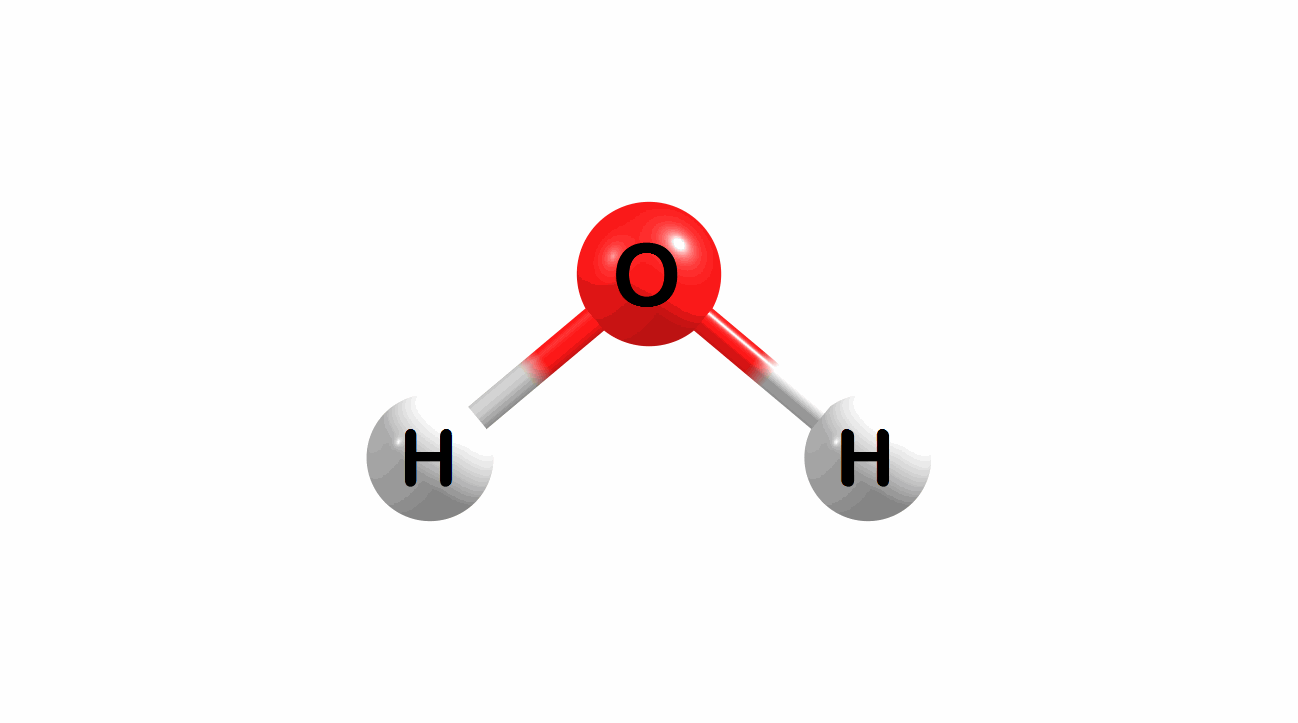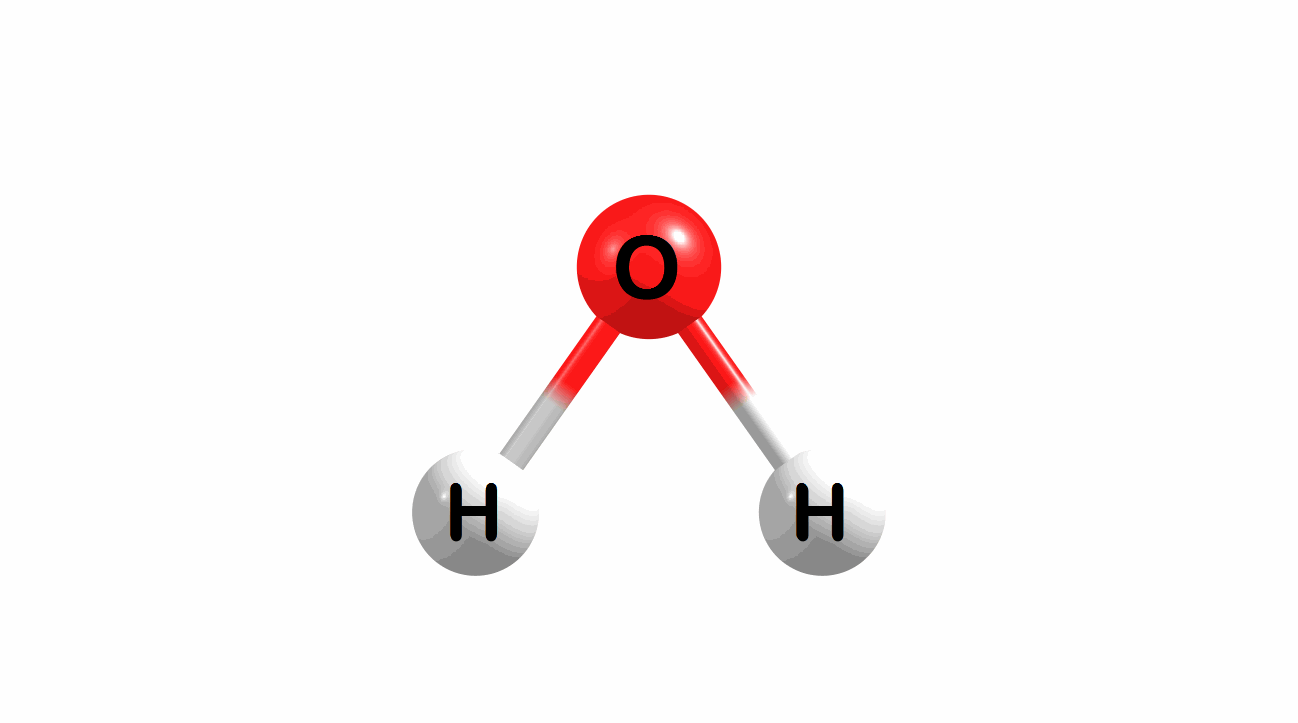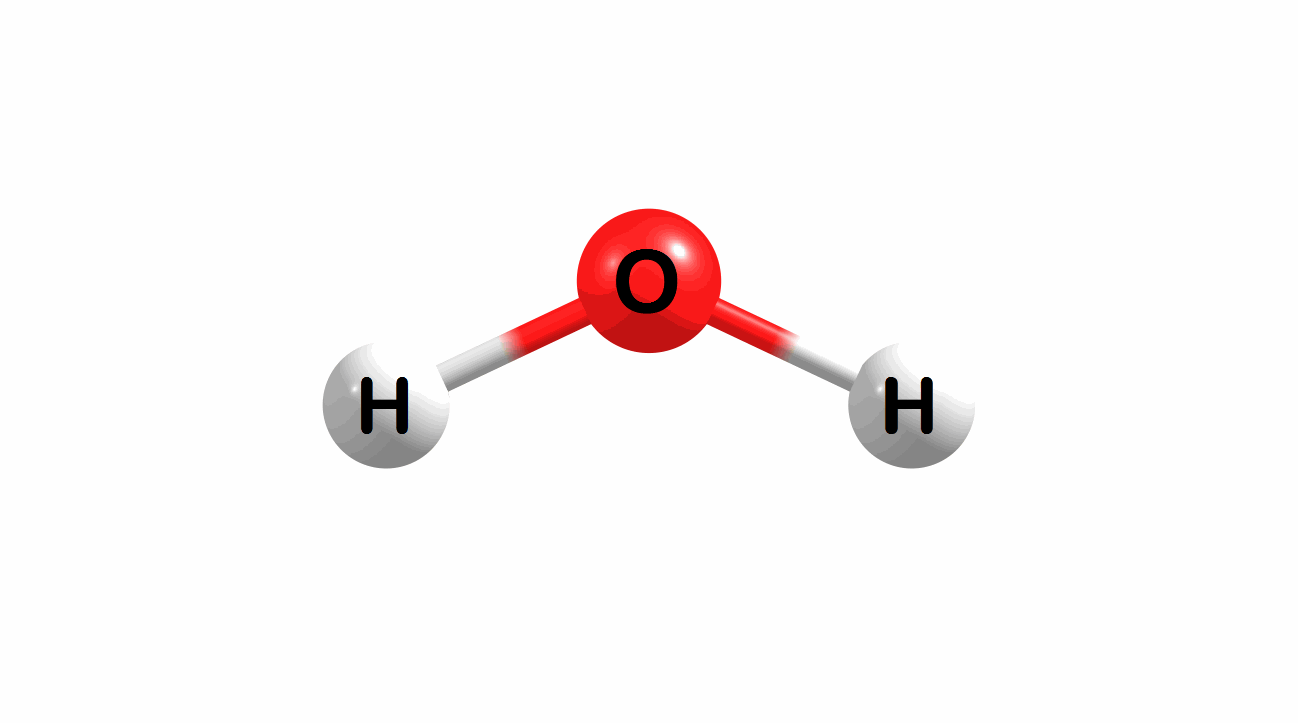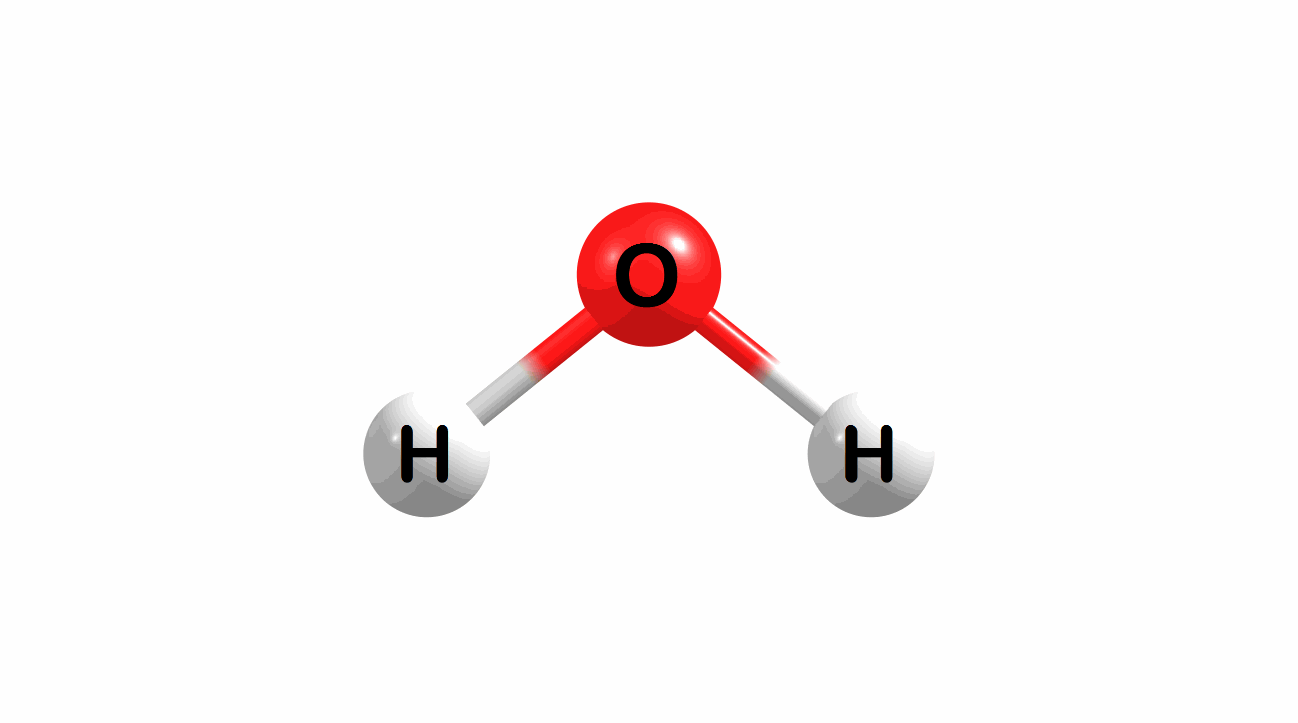
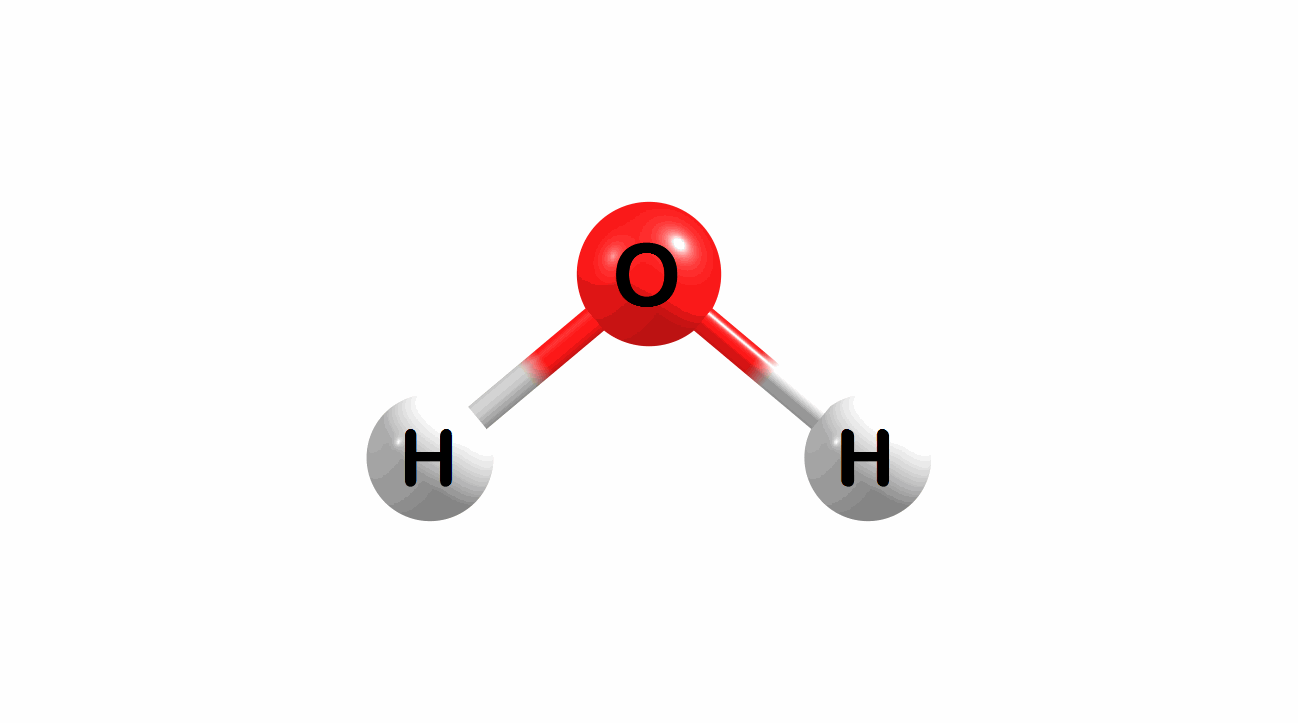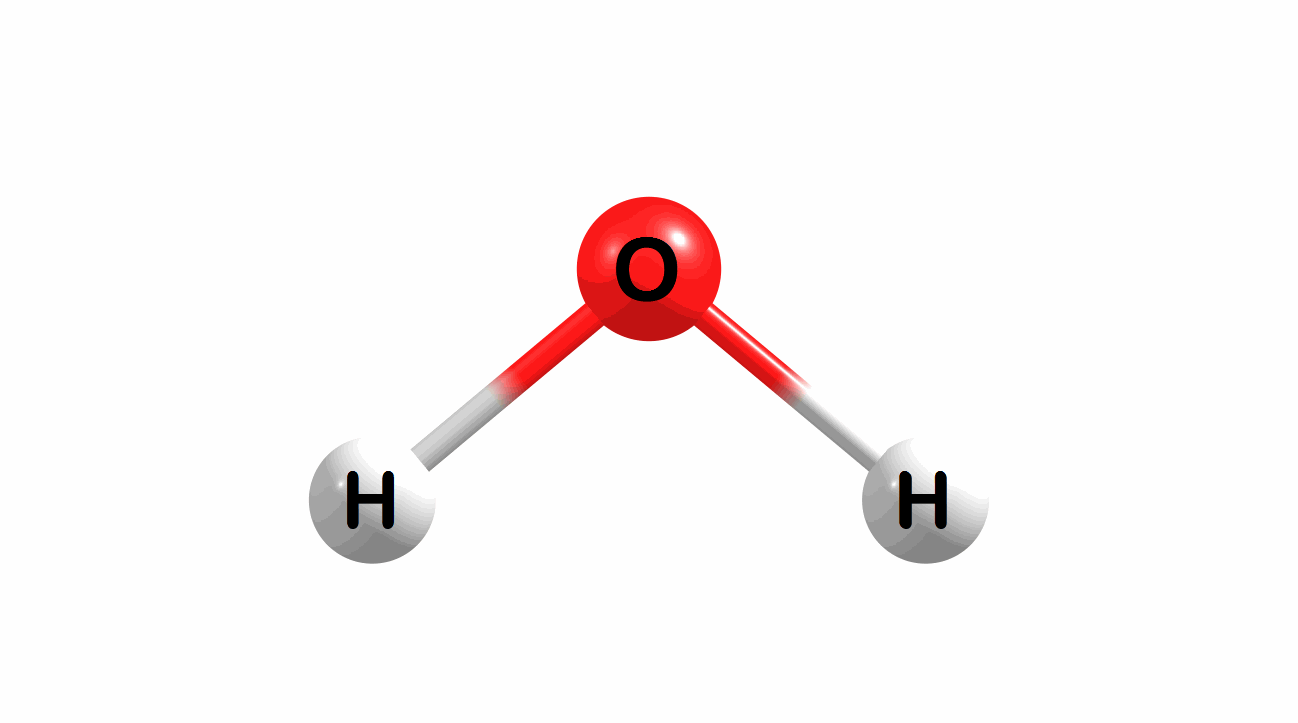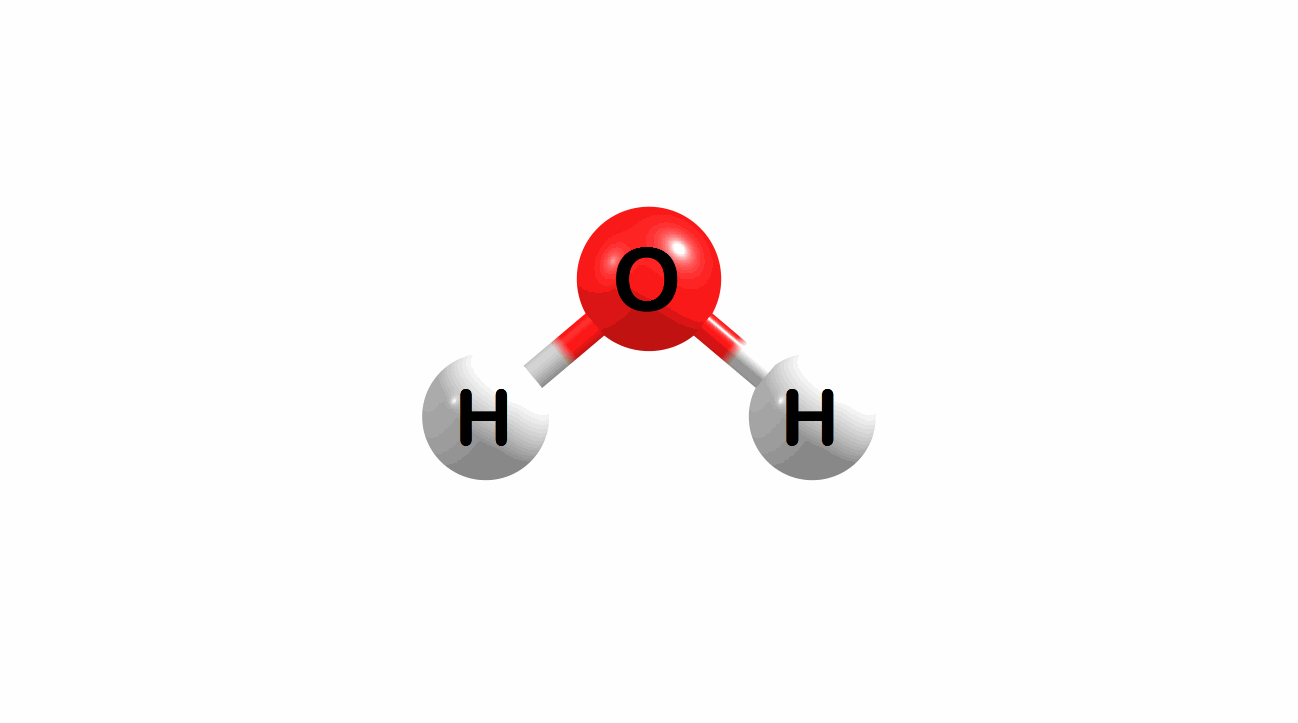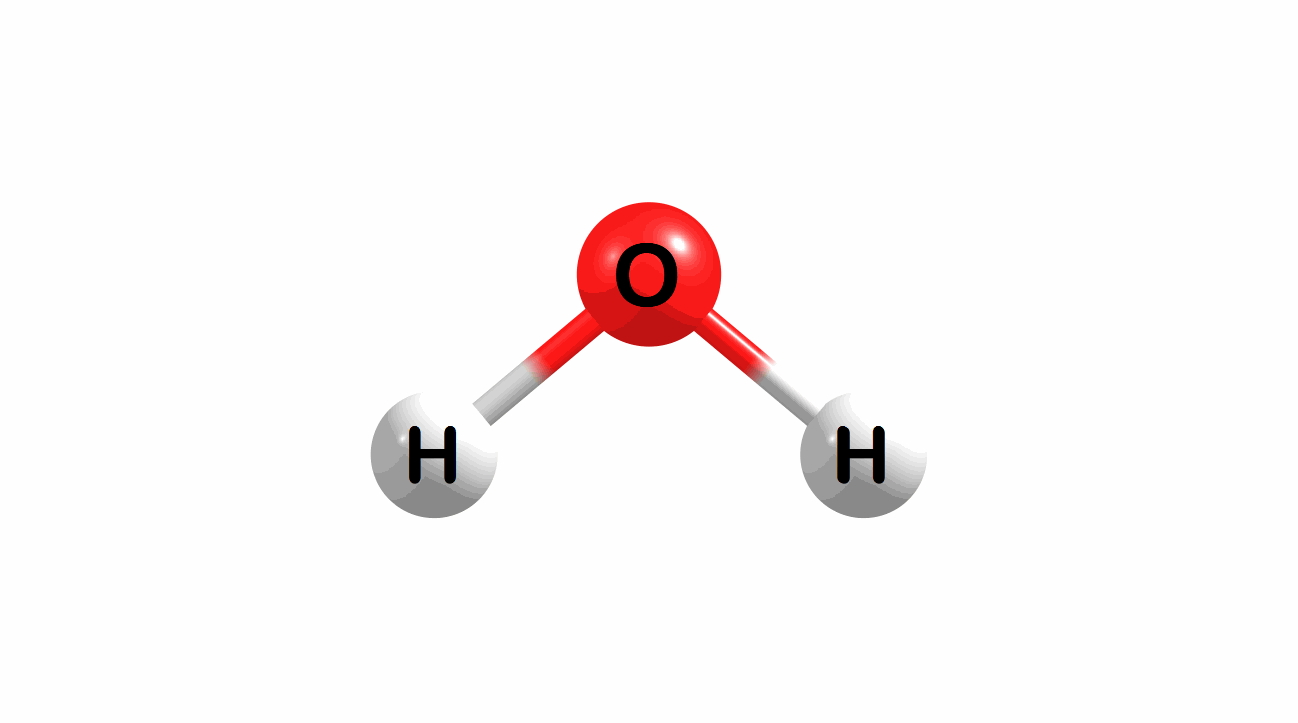
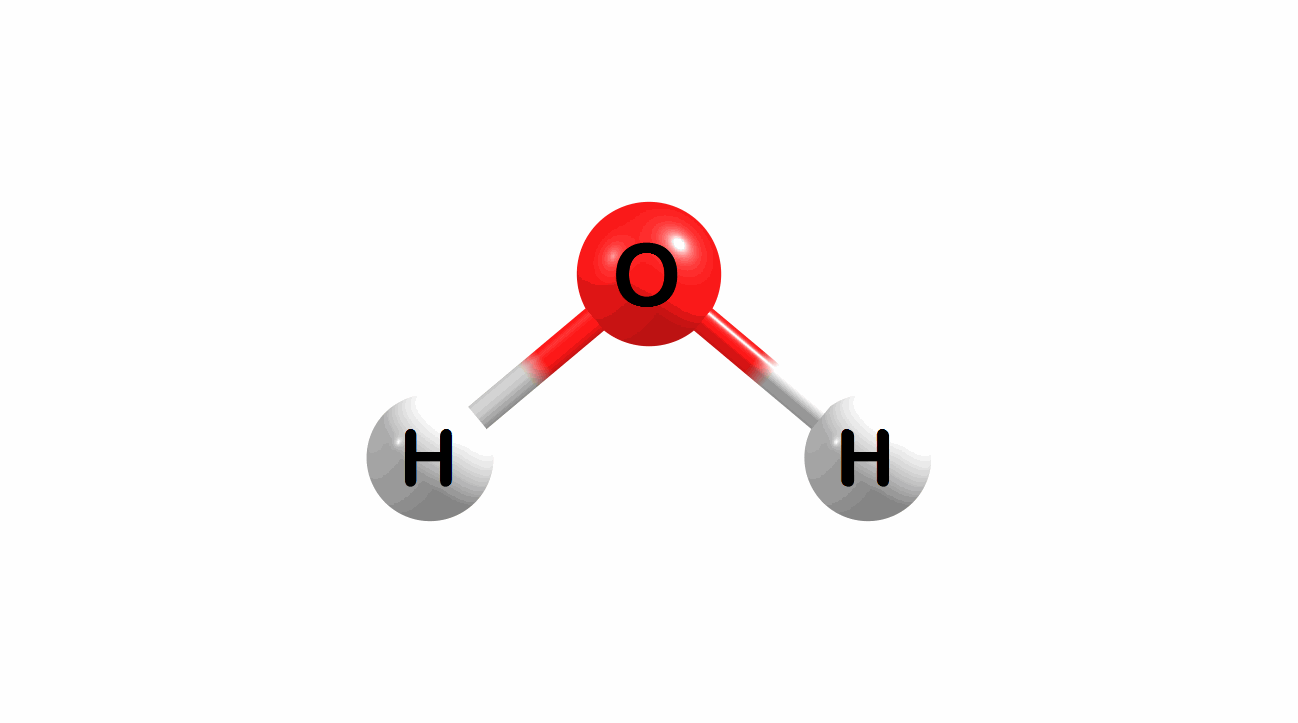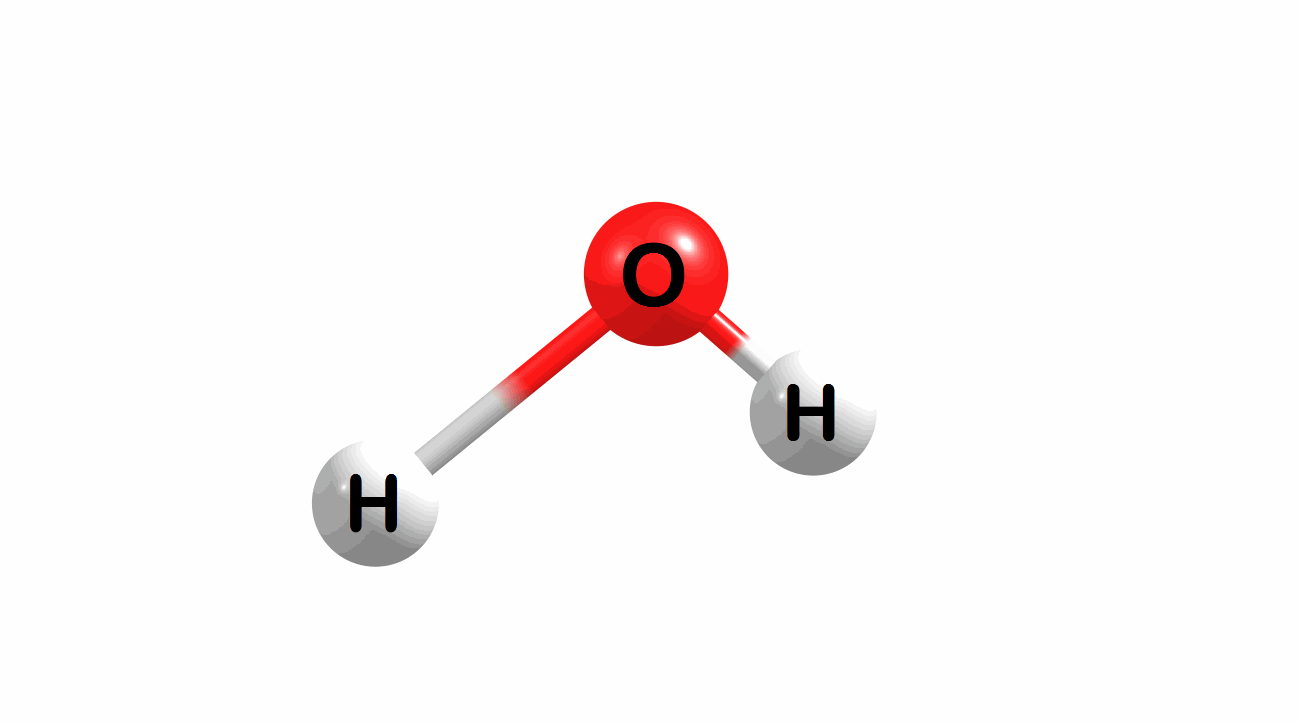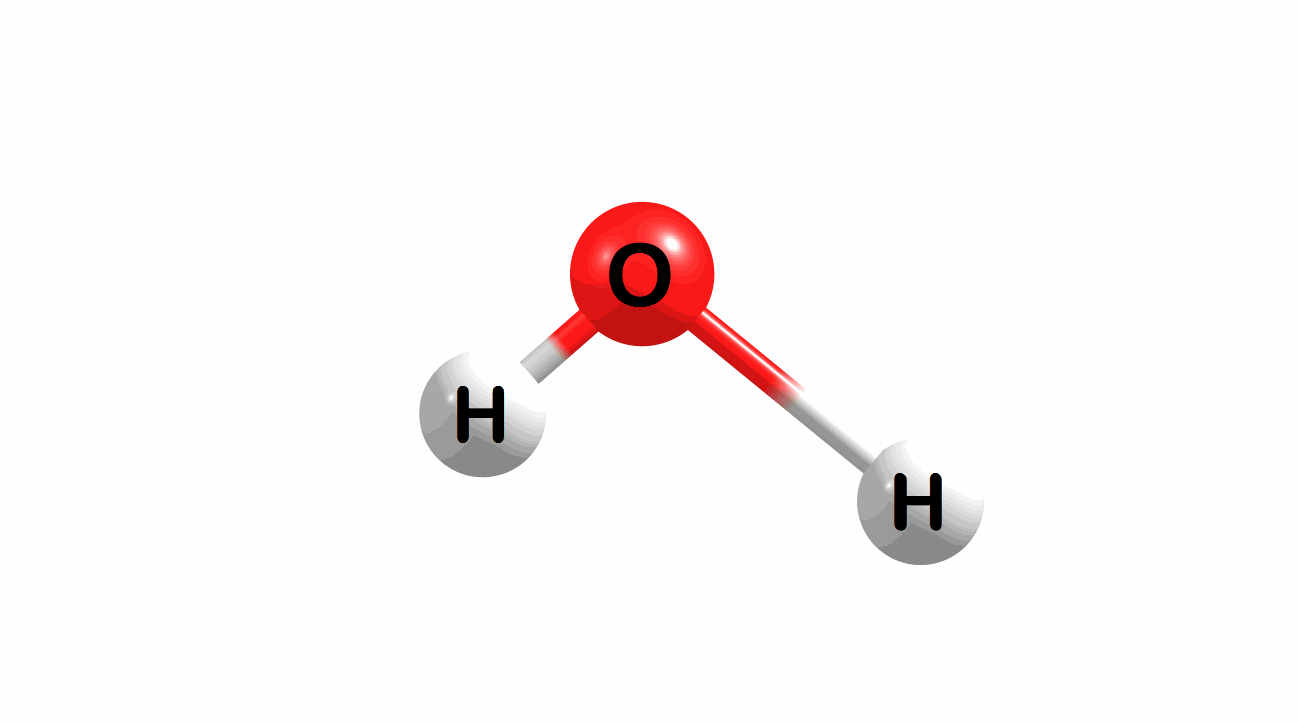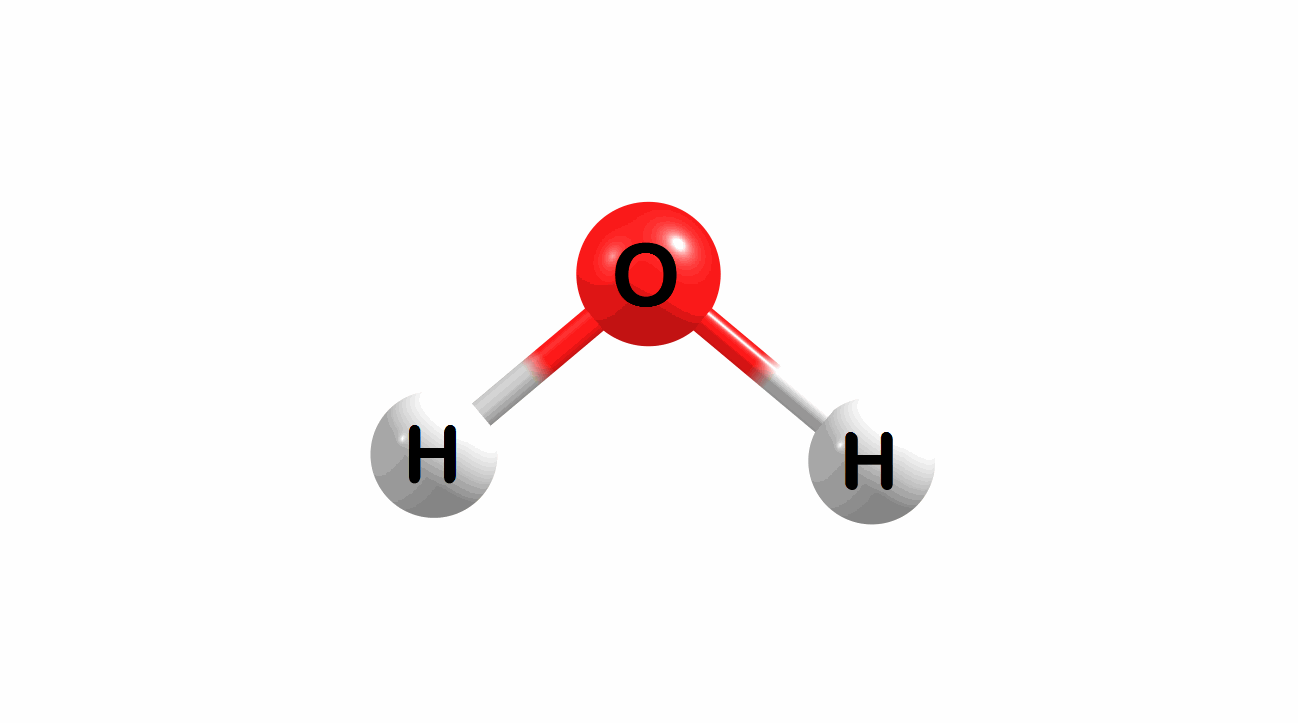
Figure 1.4: Animated fundamental vibrational modes for water.
These modes correspond (left-to-right) to the following vibrational spectra for water.
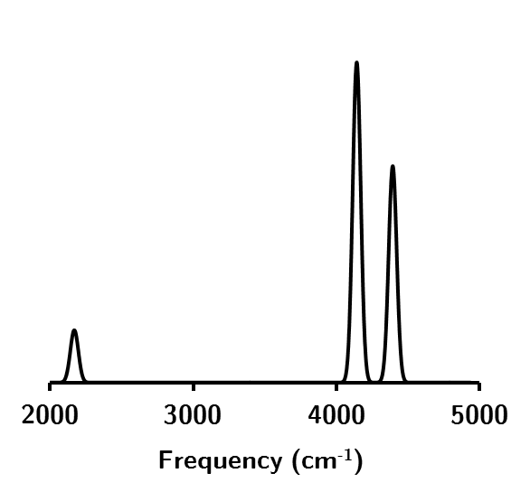
Figure 1.5: The vibrational spectra for water at the HF/STO-3G level of theory.
1.1.5 Reaction pathways
Computational chemistry can also characterize reaction pathways for chemical processes. Consider ethane, H3C-CH3. There exists a free rotation about the C-C single bond giving a “staggered” \(\longrightarrow\) “eclipsed” \(\longrightarrow\) “staggered” set of conformations.
Figure 1.6: The staggered (left and right) and eclipsed (center) conformers of ethane.
General chemistry knowledge indicates that the staggered formation should be the lower energy geometry. The rotation can be mapped as a function of energy and plotted. Below is the energy profile for this rotation.
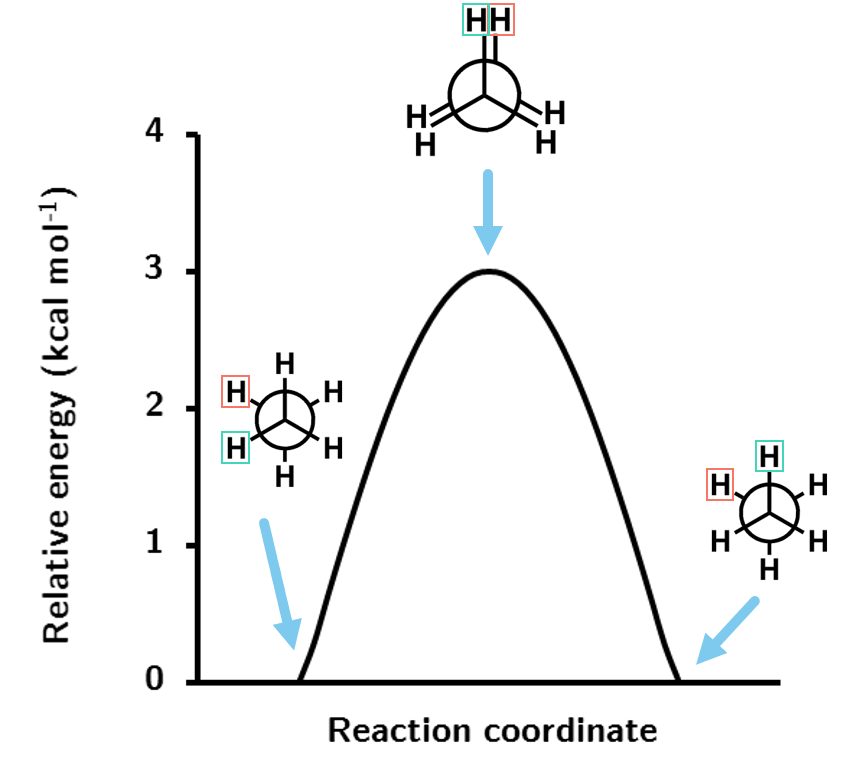
Figure 1.7: The energy profile for the rotation of ethane about the C-C bond at the HF/STO-3G level of theory.
The rotation profile shows that the energy barrier for rotation through the eclipsed conformation is just shy of 3 kcal mol-1, a very small energy barrier as expected.
1.1.6 Thermodynamic quantities
Thermodynamic quantities such as \(\Delta H\), \(\Delta S\), and \(\Delta G\) may also be computed.
1.1.7 Many more
- Excited states
- Magnetic properties (e.g. NMR)
- Emission/absorption spectra
- Dynamics
- Solvation effects
References
Connors, Kenneth Antonio. 1990. Chemical Kinetics: The Study of Reaction Rates in Solution. Wiley-VCH Verlag GmbH.